

This is the first in a series of articles about the metabolic components of adenomyosis, endometriosis, and likely several other common, but ill-understood, female reproductive conditions. In this particular post, I propose that these disease processes are driven by poor mitochondrial capacity, which in turn, is dictated by inadequate nutrient availability in key pathways responsible for the metabolism of glucose and fatty acids into ATP. The evidence comes from a variety of studies, often from disparate fields of research, pieced together to provide what I hope is a more coherent and integrative framework than what is currently available. This particular article is long, and I apologize in advance for that, but the length was necessary to provide a foundational framework for understanding my perspective.
Etiology and Pathology
Adenomyosis and endometriosis are marked by rogue endometrial cell growth, with the former confined to the myometrium and the later implanting more broadly in tissues and organs throughout the body. Adenomyosis affects anywhere from 1–20% of the female population and is present in up to 50% of women with endometriosis, while endometriosis affects approximately 10% of the female population. Given the poor diagnostic acuity with endometriosis, however, I suspect the number of affected women is much greater. Both conditions are excruciatingly painful and neither has adequate diagnostic or treatment options, largely because there is no clear understanding of cause.
Adenomyosis is believed to be predominantly iatrogenic and precipitated by surgical procedures common to women across the reproductive lifespan. A recent report, however, indicates a high incidence of adenomyosis in adolescents and young women. Absent the surgical injuries, these cases may correspond to latent endometriosis, but that too is equivocal.
The pathogenesis of endometriosis is unclear. The three prevailing theories, developed over 100 years ago, describe aspects of the disease process, but fall short of a theory that encapsulates its totality and none can be fully substantiated by evidence. More recent attempts to define genetic or epigenetic patterns associated with endometriosis, while intellectually exciting, provide neither necessary nor sufficient evidence for pathogenesis. They simply demonstrate patterns of expression associated with the disease. Ditto for the immune reactivity models.
The clinical consequences of this are broad. To date, diagnosis of either condition can take upwards of a decade. Medical treatment options are limited, largely ineffectual, and carry significant side effects. Surgery, either to remove the uterus or the aberrant tissue growth, is considered the best, and really, the only opportunity for alleviating the disease-induced pain. This is ironic, not only because surgical injury is a driving factor for the development of adenomyosis, but also, because it does nothing to address the aberrant cell growth. It simply clears the most obvious obstructions. Importantly, because the cause of these disease processes remain unknown, we have no way to prevent them.
Much like the blind men attempting to describe an elephant, we are lacking a key conceptual framework from which to identify what is before us. We have several component observations and a few theories to describe portions of disease expression, but no unifying theory that accurately explains the colossal list of potential genetic, epigenetic, hormonal and environmental ‘stressors’ capable of initiating and/or exacerbating these disease processes. I believe a mitochondrial, and thus, metabolic perspective offers that framework.
The Metabolic Factor
My work in mitochondrial metabolism and recent reports confirming that metabolism drives cell fate decisions, and thus genetic and epigenetic expression, tells me, that metabolism, is the key component of disease generally, and adenomyosis and endometriosis, more specifically. Briefly, research shows that while genetics provides a blueprint and epigenetics adds some color, metabolic capacity determines whether what is programmed in that blueprint will reach fruition, where, and in what final form. Two quotes that aptly capture this process:
The cell tests whether it has the materials in its environment. If it cannot execute the metabolism, then it won’t become that cell type, in spite of signals to differentiate.
Instead of thinking about the gene expression networks just happening to interact with metabolism, it’s really metabolism driving [developmental decision-making]…‘and gene expression networks are the tools by which that occurs.
This is a radical idea and flips the script from genetic hardwiring to a more malleable process of development; one where metabolic capacity, or lack thereof, can completely upend the form, function, and location of cells. From this perspective, aberrant cell growth, whether it be in disease processes like adenomyosis, endometriosis, or even cancer, and the accompanying and accruing genetic or epigenetic alterations, represent not something random or hardwired, but something more specific and flexible. These changes represent an active metabolic stress response. They are adaptations enacted to survive an inhospitable environment; adaptations that include emergency signals to the cell’s nucleus to chart a different path.
Why Stem Cells? Why Metabolism?
The endometrium of the female uterus sheds and regenerates itself monthly from scratch, some 400-500 times across the lifespan, all in preparation for the possibility of pregnancy. That is a remarkable feet of biological programming that many of us, myself included, have not fully appreciated. Underlying all of the hormonal, immune, physical signals that guide this process are stem cells carrying genetic and epigenetic materials that will inform cell growth and distribution to construct an appropriately pregnancy-receptive environment, or absent that pregnancy, efflux the entire thing and begin anew.
In order for this construction/deconstruction process to work properly and for those stem cells to develop as intended, they require a metabolically receptive environment with adequate nutrients and molecular substrates. This is an energetically intensive process such that any impediments will alter stem cell differentiation patterns in a manner that perfectly mirrors those observed in both adenomyosis and endometriosis.
Specifically, when a stem cell approaches division and development into its genetically recommended form, its mitochondria scan the environment. If the environment is not amenable, meaning there are not enough nutrients and other substrates needed for growth, metabolic drivers will override the original genetic and epigenetic plans to adapt accordingly. These adaptations are first and foremost protective mechanisms to ensure cell survival. The more common cell survival mechanisms include excessive proliferative and invasive behaviors along with a tendency to migrate to far off locales – exactly, the characteristic markers of adenomyosis and endometriosis.
Genetic patterns identified in the stem cells of women with adenomyosis and endometriosis confirm these patterns. Both disease processes are marked by shifts in stem cell activity. With adenomyosis, we see it in the Schwann cells and with endometriosis we see these patterns in stem cells that have migrated out of the uterus.
Stem Cell Behavior in Adenomyosis and Endometriosis
Adenomyosis is precipitated by nerve injury in a region of the uterus called the endometrial-myometrial interface or EMI. Alterations in stem cell behavior have been observed in this region, particularly in the Schwann cells. Schwann cells form the protective myelin sheath around peripheral nerves. With nerve injury, Schwann stem cells de-differentiate to their juvenile form to allow the nerve time repair. When the repair is complete, they differentiate again to mature Schwann cells and re-form the protective myelin coating. This is a hugely metabolically intensive project.
In women with adenomyosis, I believe poor metabolic capacity leaves the Schwann cells in the EMI de-differentiated and the nerves in that region unprotected. This then elicits severe, neuropathic pain, along with increased and persistent inflammation and dysregulated immune function. Importantly, the inability to heal from this injury is what drives, not only the problems with the Schwann stem cells, but the proliferation and migration of other stem cells (those responsible for the monthly shedding and rebuilding the endometrial lining) into the myometrium. This has been demonstrated experimentally.
Neuropathic pain is common to women with endometriosis as well, even absent evidence of physical or surgical injury. This suggests an underlying metabolic derangement favorable to uterine and other neuropathies. In endometriosis, however, the problems with stem cell metabolism are broader, more deeply skewed, and likely generationally entrenched; something we will talk about more in a moment. For now, however, understand that endometriotic stem cells exhibit the same adaptations linked to poor metabolism as those identified experimentally in other cell populations.
Stem cells isolated from endometriosis tissue exhibited increased migration and proliferative capability during in vitro assays compared to eutopic endometrium stem cells, and transplanted MSCs isolated from ectopic endometrial tissue demonstrated higher levels of invasiveness and vasculogenic potential.
Indeed, like the persistently juvenile stem cells observed with adenomyosis:
Ectopic endometriosis cells respire less at rest and have reduced max respiratory capacity … [and] have more stem-cell like properties than their eutopic counterparts.
Reduced respiratory capacity is the key here. Respiration denotes the cell’s ability, or more specifically, the capacity of the mitochondria within that cell, to utilize oxygen and nutrients to produce ATP. Juvenile or de-differentiated stem cells rely on glycolysis for energy and substrates. So too do cancer cells and this is what drives tumorogenesis. In order for the stem cell to become appropriately differentiated into their recommended form and not to replicate wildly, it has to switch over to mitochondrial metabolism for energetics. This appears not to be happening, at least not efficiently or consistently, and it is what we need to address with adenomyosis and endometriosis.
What is Metabolism?
Before digging more deeply into the stem cell/metabolism interactions, it is important to define some terms and concepts. Namely, we need to understand what metabolism is and what drives it. Briefly, metabolism is how the body pushes nutrients through a variety of enzymatic reactions, both in the cell and the mitochondria, combines it with oxygen, and makes energy or ATP and all of the other molecules required for cell functioning, cell survival, and managed apoptosis. The degree to which this is done efficiently is considered metabolic capacity; the degree to which this is done efficiently in the face of illness, injury, or stressor, is metabolic resilience. Both play a role in the onset and persistence of disease. One can have apparently sufficient capacity in a steady state, but poor resilience, and thus, decline rapidly when exposed to a stressor, even a mild one. In some cases, this represents a compensated capacity; a state where absolute capacity has waned but the body successfully compensates by enacting of survival mechanisms that mask deficiencies. Short term, these mechanisms are protective, long term, however, they reduce resources even further, escalate and entrench illness patterns marked by increasing inflammation, immune reactivity, and dysregulated hormone patterns; hallmarks of adenomyosis, endometriosis, and a long list of other diseases processes.
I should note, that metabolism also includes the manner and efficiency by which cells process and clear toxins. This aspect is what most of us were taught to focus on, especially in the context of pharmaceutical medicine where it is important to understand how drugs are processed through the body. This is a secondary function, however, in the sense that it is entirely dependent upon mitochondrial energy production and substrate availability, which in turn is dependent on nutrient availability. Poor energy and limited substrate availability equals poor detoxification even in the face of minimal toxicant exposure.
Metabolism is driven by environment, in its totality. Everything we eat, drink, are exposed to, and do, and everything our parents and grandparents and on, ate/drank, were exposed to, and did, shapes metabolic capacity and resilience. The genetic component of metabolism is the memory of their toxicant exposures and nutrient intake struggles expressed or not expressed by our exposures and struggles. Some think this is epigenetics, the soft-coded reregulation of genetic activity. I believe it is more fundamental, and ultimately, more malleable, because except in rare circumstances, one’s inherited or baseline metabolic capacity and resilience is continuously modifiable, both positively and negatively, by the current environment.
Mitochondria Control Metabolism
At the hub of metabolism, sit the mitochondria. These are the organelles within each cell that are responsible for integrating vast environmental signals and matching the needs of the cell with the energy and other supplies requisite for survival. It is their job to keep the cell alive. To that end, mitochondria produce >90% of the ATP required for cell functioning. Limit that capacity, and all sorts of things go wrong.
Mitochondrial capacity is contingent on nutrient intake; both macronutrients, as in protein, carbohydrates and fats, and micronutrients, as in vitamins, minerals, and metals. In order for consumed macronutrients to become ATP, and this is the key point that most discussions of nutrition and mitochondrial functioning miss, there must be adequate intake of micronutrients (and what defines adequate is relative to the individual’s unique needs). Micronutrients are cofactors in all of the enzymes responsible for metabolizing food into ATP and into many other molecules that dictate cell behavior. This means that inadequate micronutrient availability will reduce ATP output, regardless of macronutrient consumption. Figure 1., below, illustrates the mitochondrial nutrients required to produce ATP.
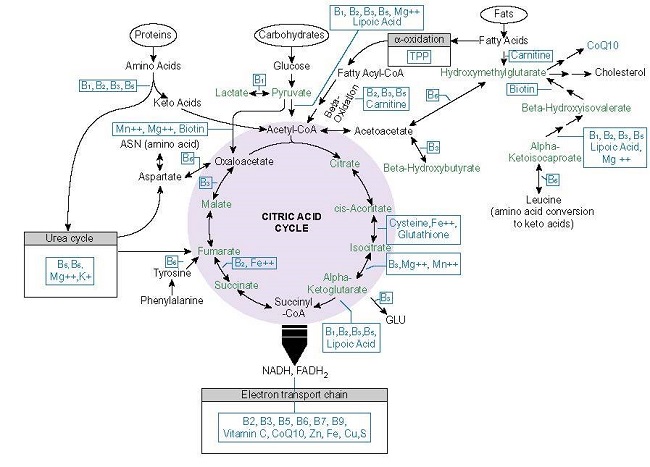
So, when we speak of metabolism, think mitochondria and think nutrients, and when we think of nutrients, think of the vitamins, minerals, and metals that power the enzymatic reactions required to produce energy from macronutrients. Inadequate micronutrient intake diminishes mitochondrial/metabolic capacity by altering enzyme activity. This reduces resilience and initiates survival compensations; survival compensations that come at an energetic cost, which carried across time and even generations, alter stem cell behavior, particularly in the reproductive stem cells that were fostered by the experiences of our grandmothers and great-grandmothers.
The Energy Crisis in Adenomyosis and Endometriosis
Evidence of poor mitochondrial capacity in adenomyosis and endometriosis comes from multiple sources including research looking at the structure of mitochondria within the lesions, their behavior or dynamics, a growing number of ‘omics’ studies, as well as more direct assessment of mitochondrial respiration itself. Specifically, research shows that the mitochondria within lesions are deformed in manner indicative poor energetic capacity. As a consequence, how they interact with each other and navigate life and death cycles, is also disturbed, as is their ability to respire – e.g. use oxygen and nutrients to produce energy.
Why the mitochondria are deformed, behave aberrantly, and respire less, has everything to do with nutrient availability, which in turn is modified both by intake and toxicant exposure (toxicants deplete nutrients, damage mitochondria, and reduce metabolic capacity). Additionally, there is evidence of metabolic reprogramming consistent with poor mitochondrial energetics and a clear shift away from mitochondrial energy production, towards the Warburg-like metabolism patterns noted in cancer cells. Finally, direct measurement of mitochondrial respiration confirm reduced oxidative/energetic output.
The question is why? Why is mitochondrial metabolism impaired? The answer rests with nutrient > enzyme interactions. There are two key patterns, repeated across studies (here, here, here), that I believe account for much of the aberrant mitochondrial metabolism and resultant changes in cell and stem cell behavior in both of these disease processes. I should note, however, that most the research involves endometriosis cells, as there is a general paucity of omics research addressing adenomyosis specifically.
Despite this, the patterns identified link directly to deficiencies in specific nutrients, one in particular, but possibly two. The first pattern involves the interaction between two thiamine (vitamin B1) dependent enzymes, the pyruvate dehydrogenase complex (PDHC) and the pyruvate dehydrogenase kinase (PDK). The second pattern involves alterations in something called the carnitine shuttle and the subsequent activation of ketogenic metabolism. The carnitine shuttle is influenced by thiamine status, but it is also dependent upon carnitine (a substance derived from the consumption of meats). The pathways that these enzymes control are responsible for ~90% of the total mitochondrial energy production and both are disturbed in endometriosis, and likely also adenomyosis as well.
In the first pathway, the PDHC is blocked by the PDK. The PDHC controls glucose metabolism, determining whether the process stops at glycolysis yielding only 2 molecules of ATP per molecule of glucose or continues on through the mitochondria to yield up to 32 molecules. The PDHC has three subunits and each require a nutrient cofactor to function. The first subunit is dependent upon thiamine and second and third are dependent upon lipoic acid and riboflavin, respectively. While deficiency in any of these cofactors is problematic, as a cofactor to the first subunit in this enzyme, thiamine is absolutely critical. Its absence effectively shuts the glucose pathway down, reducing mitochondrial energetics significantly. PDK blocks the thiamine dependent subunit of the PDHC enzyme and it is upregulated in endometriotic cells.
The PDK enzyme is metabolic sensor that slows mitochondrial energy production when nutrient cofactors are low and/or when toxicants are high. It does so by shifting energy production from the mitochondria (~32 ATP) to the cell (2 ATP), while upregulating the compensatory energetic pathways involving increased protein synthesis, cell migration, cell growth, differentiation, and proliferation – e.g. the cancer-like properties of adenomyosis and endometriosis.
To that end, the PDK enzyme (there are actually 4 shapes or isoforms of the enzyme that reside preferentially in different tissues), is upregulated by thiamine deficiency and downregulated by thiamine sufficiency. It is also upregulated by a long list of toxicants via multiple mechanisms, beyond the scope of this article, but which I will cover in a subsequent post. Briefly, however, several environmental chemicals and some pharmaceuticals (PPAR agonists), food additives (seed oils, among others), plastics, and other nutritional and metabolic compounds, like vitamin A (deficiency upregulates) and melatonin (deficiency upregulates) modulate PDK activity.
The second key energetic pathway altered in women with adenomyosis and endometriosis is the fatty acid pathway. A single molecule of fat, when metabolized appropriately in the cell and again in the mitochondria, can yield ~ 100 molecules of ATP, making this a major energy pathway, particularly in muscle tissue, whether it be in the heart, the skeletal muscle, or the myometrium.
Unlike the evidence of poor glucose metabolism/energetics, which is quite clear, evidence of disturbed fatty metabolism is more complicated and somewhat equivocal. Nevertheless, I believe this pathway disrupted as well, both by the lack of thiamine and a potential lack of carnitine.
Fatty acid metabolism goes through multiple stages of processing before arriving at the mitochondria. The first stage occurs in a set of organelles called peroxisomes. This stage is called alpha oxidation and it is where another thiamine dependent enzyme called the HACL-1 lives. HACL-1 is responsible for reducing long chains of fatty acids to shorter chains, but its activity is reduced in the absence of thiamine. Given the pattern of upregulated PDK activity and reduced mitochondrial respiration more generally, we can surmise that thiamine is inadequate and that there is likely a bottleneck in the alpha oxidation component of fatty acid metabolism.
Further down this pathway, live the carnitine shuttles, which move the product of reduced fatty acids (acyl-CoA) into the mitochondria so that they can be processed and eventually become ATP. The first of the two carnitine shuttles (which moves the product across the mitochondrial membrane) is upregulated. This is where it gets confusing. Normally, upregulation of a key transporter would indicate that more product is metabolized into energy. I believe this pattern suggests the opposite, however. Let me explain why.
Thiamine, in addition to being a requisite cofactor in the PDHC, the PDK, and the HACL (and the a-ketoglutarate dehydrogenase enzyme within the mitochondria, and transketolase enzymes within the cell), is also a rate-limiting cofactor in an enzyme responsible for the metabolism branched chain amino acids (BCAA: the valine, leucine, and isoleucine components of protein). This enzyme is called the branched chain keto acid dehydrogenase (BCKAD). When thiamine is low, catabolism of these amino acids is incomplete and produces a surplus of potentially toxic compounds called acylcarnitines. These are the same acylcarnitines from fatty acid metabolism that must use the carnitine shuttle for transfer into the mitochondria and the eventual processing into ATP.
I believe the excess acylcarnitines from the thiamine-starved BCAA pathway are effectively overwhelming the carnitine shuttle, driving its upregulation as a compensatory reaction and that this change is indicative of increased metabolic stress not increased energy production. Supporting this hypothesis are 1) are consistently noted reduced mitochondrial respiration discussed throughout this article, and 2) the increase in ketones absent conscious dietary changes to induce ketogenesis, observed in other studies. Ketones are a source of rescue energy that are metabolized in the face of glucose starvation, intentionally with ketogenic diets, or unintentionally, as I suspect is the case here, when glucose metabolism fails.
From the Mitochondria to the Cell
Presented above are the clues that I have uncovered thus far linking derangements in mitochondrial energy production to to cell behavior. I am building a case to argue that metabolism, or more specifically, reduced metabolic/mitochondrial capacity drives the aberrant cell behavior observed in these disease processes, which in turn drives the expression of genetic and epigenetic proteins to compensate. Admittedly, much of this hypothetical and draws on pieces of research from somewhat disparate sources, and I have not fully elucidated several key components of this theory, but I believe this model offers not only a more accurate framework for understanding, but it affords us an opportunity to prevent, and more effectively, treat these disease processes than does the genes first model currently employed. For if metabolism is the driver of genetic expression, and if metabolism is affected by nutrient availability and toxicant exposures, those variables are entirely correctable.
In subsequent posts, I will continue to unpack mechanisms and more fully layout the metabolic drivers of adenomyosis, endometriosis, and other female reproductive disease processes. For now though, I encourage readers to investigate their own metabolic/mitochondrial/nutrient weaknesses. We have hundreds of articles on these topics.
I will leave you with this final quote.
…alterations in mitochondrial function precede changes in transcription that are deterministic for stem cell fate.
We Need Your Help
More people than ever are reading Hormones Matter, a testament to the need for independent voices in health and medicine. We are not funded and accept limited advertising. Unlike many health sites, we don’t force you to purchase a subscription. We believe health information should be open to all. If you read Hormones Matter, like it, please help support it. Contribute now.
Yes, I would like to support Hormones Matter.
Photo by Stephen Harlan on Unsplash
Part 1.5 in the series: Environmentally Upregulated PDK Blocks Key Thiamine Dependent Enzyme













